计算任务提交(入门版)
本节内容将介绍如何在 SonmiHPC 集群上提交你的第一个计算任务。以下的操作前提是你已经通过终端工具登录连接上了集群。
我们以提交一个简单的 LAMMPS 分子动力学计算为例,假如你对该计算软件或者领域没太多了解那也不妨碍继续下面的教程,因为在各个计算学科领域的这种计算软件都类似。并且集群中也已经提供了预编译好的 LAMMPS 并行程序。
创建项目目录
通过以下命令进入自己的家目录中,并创建一个项目目录lammps,进入项目目录中:
[sonmi@sonmi ~]$ cd ~
[sonmi@sonmi ~]$ mkdir lammps
[sonmi@sonmi ~]$ cd lammps[sonmi@sonmi ~]$ cd ~
[sonmi@sonmi ~]$ mkdir lammps
[sonmi@sonmi ~]$ cd lammps准备LAMMPS的计算输入文件
LAMMPS的程序需要读取计算文件,作为简单的例子我们的输入文件也比较简单。在项目目录下面使用vim或者其他编辑器创建lj.in文件:
# 3d Lennard-Jones melt
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 20 0 20 0 20
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 1.44 87287 loop geom
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify delay 5 every 1
fix 1 all nve
run 20000# 3d Lennard-Jones melt
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 20 0 20 0 20
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 1.44 87287 loop geom
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify delay 5 every 1
fix 1 all nve
run 20000准备SLURM的批处理提交脚本
我们需要将计算任务提交到slurm队列系统中。在项目目录下使用vim或者其他编辑器创建lammps.job文件:
#!/usr/bin/bash
#SBATCH --job-name=lmp # 自定义任务名称
#SBATCH --partition=sonmi # 想将任务分配到的节点分区
#SBATCH --output=output.log # 任务计算过程标准输出文件
#SBATCH --error=error.log # 任务计算过程错误输出文件
#SBATCH --nodelist=compute-0-[0-1] # 指定任务提交的计算节点
#SBATCH -n 8 # 指定任务需要占用的CPU核数
ulimit -s unlimited
ulimit -l unlimited
cd $SLURM_SUBMIT_DIR
source /opt/intel/oneapi/setvars.sh # 激活计算程序需要用到的环境变量
mpirun -np 8 lmp -in lj.in -l lmp.log # lmp为LAMMPS程序,-in 参数指定读取的输入文件,mpirun使用消息传递接口可以进行跨节点并行计算#!/usr/bin/bash
#SBATCH --job-name=lmp # 自定义任务名称
#SBATCH --partition=sonmi # 想将任务分配到的节点分区
#SBATCH --output=output.log # 任务计算过程标准输出文件
#SBATCH --error=error.log # 任务计算过程错误输出文件
#SBATCH --nodelist=compute-0-[0-1] # 指定任务提交的计算节点
#SBATCH -n 8 # 指定任务需要占用的CPU核数
ulimit -s unlimited
ulimit -l unlimited
cd $SLURM_SUBMIT_DIR
source /opt/intel/oneapi/setvars.sh # 激活计算程序需要用到的环境变量
mpirun -np 8 lmp -in lj.in -l lmp.log # lmp为LAMMPS程序,-in 参数指定读取的输入文件,mpirun使用消息传递接口可以进行跨节点并行计算提交计算任务
在准备好计算文件跟任务提交脚本之后,就可以通过以下的命令提交计算任务了:
sbatch lammps.jobsbatch lammps.job提交之后会返回对应的任务ID。
在任务提交之后我们可以通过以下的命令查看计算任务的进度:
sonmictl job infosonmictl job info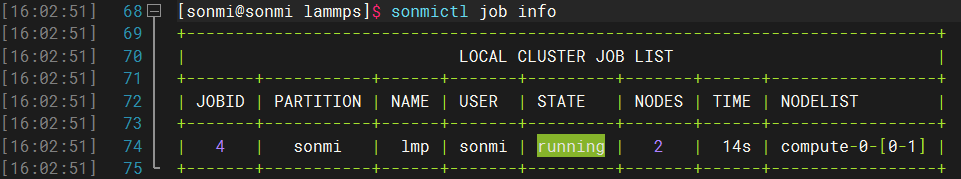
然后等待任务计算完成即可。
取消正在排队/计算的任务
当计算任务已经提交之后,无论是否已经在计算或者在排队等待中,都可以随时取消。可以使用以下命令取消对应的任务:
scancel <JOBID> # JOBID为提交任务后分配的任务IDscancel <JOBID> # JOBID为提交任务后分配的任务ID